If this study has captured anything beyond systematically correlated measurement errors (a problem which plagues high-throughput sequencing assays), it is almost certainly ancillary population substructure which correlates with socio-economic status.
Alex, why do you say these hurtful things? You know perfectly well that they use principal components to successfully control away population structure, that the genetic correlations between cohorts are high, the hits replicate across populations, the enrichment sets and tissue expression are predominantly in the nervous system as expected, the polygenic scores are predictive of success even looking at just kids in poor families, and the within-family polygenic scores perform fine. No, it's not some measurement error on the several different SNP chips they used in the ~12 different cohorts or the entirely separate GWASes whose results they replicated, and it's definitely not 'residual population structure'. It was silly 4 years ago to write off GWAS results with that sort of logic, and it has only gotten sillier.
>use principal components to successfully control away population structure
PCA doesn't perfectly control away population structure, especially if you have cryptic tiny subpopulations in your dataset that only few individuals are in. In that case you don't have enough signal to establish the subpopulations. It's also a function of the way you call your SNPs - if you don't have enough SNPs, or focus on a different method, PCA won't magically know about those unobserved populations.
They may not perfectly control, but they do the job well enough that the error from that is minimal and of less importance than other issues like limited SNP panel coverage, sample error, or measurement error (a _huge_ limit on current GWASes and very underappreciated, given how few papers attempt to correct for it or even mention what sort of psychometric properties the available data has). It is certainly not true, as Alex claims, that the results are nothing but systematic measurement error or population structure.
Wow, I actually have no idea if gwern was being sarcastic or not. I didn't consider it until I read your comment. There's a few clues in there that do suggest sarcasm, but I really can't tell, because I know nothing about the field.
I don't think they were being sarcastic though, because the rest of their replies are pretty serious.
He says some things with perfect confidence which are unknowable or false, but you do need a background in the field to see that. Also, he starts out with what seems to be an outrageous appeal to emotion, but I suppose "hurtful" could mean "harmful" rather than "mean."
Nothing that special. You take the SNPs, extract the first 7 or whatever principal components, and include them in the regression. To the extent that there's some population structure and subgroups are smarter/richer, it'll get controlled away. No guarantee that it'll work, but it apparently does (probably because most of the structure is in the first few components and including PCs beyond that is harmless).
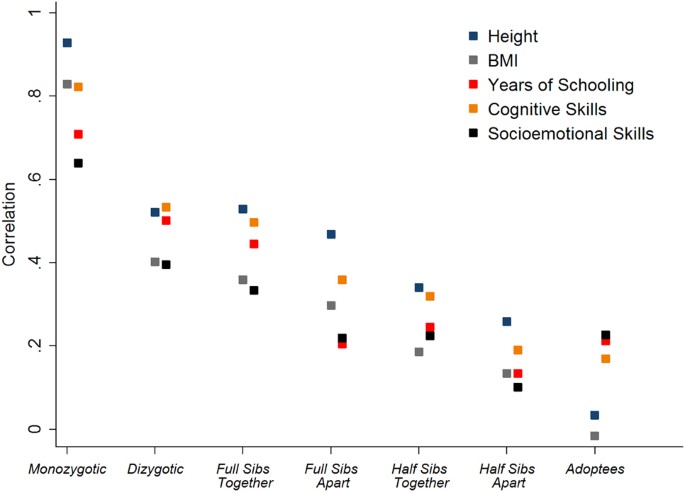
Somewhere around 70-80% of variance. People citing 50% as being conservative and generally ignoring measurement error and the heritability increasing with age. The latest GCTA methods, expanding the genetics considered but still far short of all genetic effects, turn in a lower bound of ~50% ("Genomic analysis of family data reveals additional genetic effects on intelligence and personality" http://biorxiv.org/content/early/2017/02/06/106203 , Hill et al 2017; "Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits" http://biorxiv.org/content/early/2017/03/10/115527 , Evans et al 2017), so the true figure is definitely going to be much higher.
Researchers have conducted many studies to look for genes that influence intelligence. Many of these studies have focused on similarities and differences in IQ within families, particularly looking at adopted children and twins. These studies suggest that genetic factors underlie about 50 percent of the difference in intelligence among individuals.
Well, if you compare any human child with a snail you'll find that the human does almost infinity better at answering questions on IQ tests so we could say it's all genetic. Or we could take a sample of humans and feed the control group but not the other group over a course of a few weeks and see that environment plays a huge role. But over the range of upbringings we see in the US and genotypes we see in the US it's more genetic than not. But that doesn't mean that that's true across the globe. If some dirt farmer from a place with no iodine in the salt, no folate in the bread, and no access to modern public health, medicine, or schooling comes to the US their kids will tend to have an IQ something like 20 points higher.
It's a politically-charged question, so people tend to be very careful to ensure that whatever they say is correct. It's probably difficult to give a convincing argument in the space of an average HN comment.
No, of course they have predictive value. If they didn't, that would not support my point that the GWAS results are valid. The specific study I had in mind was "The Genetics of Success: How Single-Nucleotide Polymorphisms Associated With Educational Attainment Relate to Life-Course Development" https://www.gwern.net/docs/genetics/2016-belsky.pdf , Belsky et al 2016.
I apologize for misinterpreting your response as sarcastic.
Why do you believe that PCA successfully captures all the relevant population structure? Probablistically, it's a Gaussian linear model... no reason to expect allele frequencies to follow that. If that were the case, methods like coalescent theory would be unnecessary.
Why do you believe that high genetic correlations between cohorts is sufficient to ensure safe inference from GWAS meta analysis? These are tiny effect sizes, and the argument is essentially that the correlations with intelligence measurements are statistically significant. Unless you believe that your null model is an otherwise entirely accurate reflection of how the data is generated, of course you're going to see spuriously significant results in huge samples.
I'll respond to your other points when/if I catch up on the literature. I'd be grateful if you could link the papers about the enrichment sets and tissue expression results.
{kind=link}